If you’re preparing for the United States Medical Licensing Examination® (USMLE®) Step 1 exam, you might want to know which questions are most often missed by test-prep takers. Check out this example from Kaplan Medical, and read an expert explanation of the answer. Also check out all posts in this series.
This month’s stumper
A 6-month-old boy is brought to the physician by his parents, who are first cousins, for a well-child examination. The boy appears thin, small and lethargic. Physical examination shows slightly misshapen long bones. Joint movements are restricted, the corneas are clouded, and the gums are hyperplastic. Laboratory studies of serum show elevated levels of acid hydrolases and glycosylases.
Which of the following metabolic activities is most likely defective in this patient?
A. Accumulation of gangliosides.
B. Degradation of dermatan sulfate and heparan sulfate.
C. Degradation of glycogen.
D. Degradation of sphingomyelin.
E. Phosphorylation of mannose at lysosomal-targeting sites.
F. Phosphorylation of tyrosine kinase receptors.
(Scroll down for answer.)
The correct answer is E.
Kaplan Medical explains why
This patient has I-cell disease, also known as mucolipidosis II, which is caused by a defective UDP-N-acetylglucosamine-1-phosphotransferase, the enzyme that phosphorylates a terminal mannose moiety on enzymes destined for inclusion in lysosomes. The resulting mannose-6-phosphate residue interacts with a specific receptor on the lysosomal membrane and triggers internalization. In the absence of mannose phosphorylation, the enzyme is secreted into the extracellular space.
Proteins coded by nuclear DNA are synthesized on cytoplasmic ribosomes, which may be either "free" or associated with the endoplasmic reticulum to form the rough endoplasmic reticulum (RER). Proteins synthesized in the RER are transferred into the Golgi apparatus, where they undergo further modifications that determine whether they remain part of the Golgi apparatus, become part of the plasma membrane, or are shipped to lysosomes or other locations.
Proteins not marked for transport to a specific intracellular site follow the default pathway and are exported into the extracellular compartment. Lysosomes deprived of critical enzymes (e.g., due to I-cell disease) are unable to degrade their normal substrates, the latter of which gradually accumulate and transform the lysosomes into inclusion bodies.
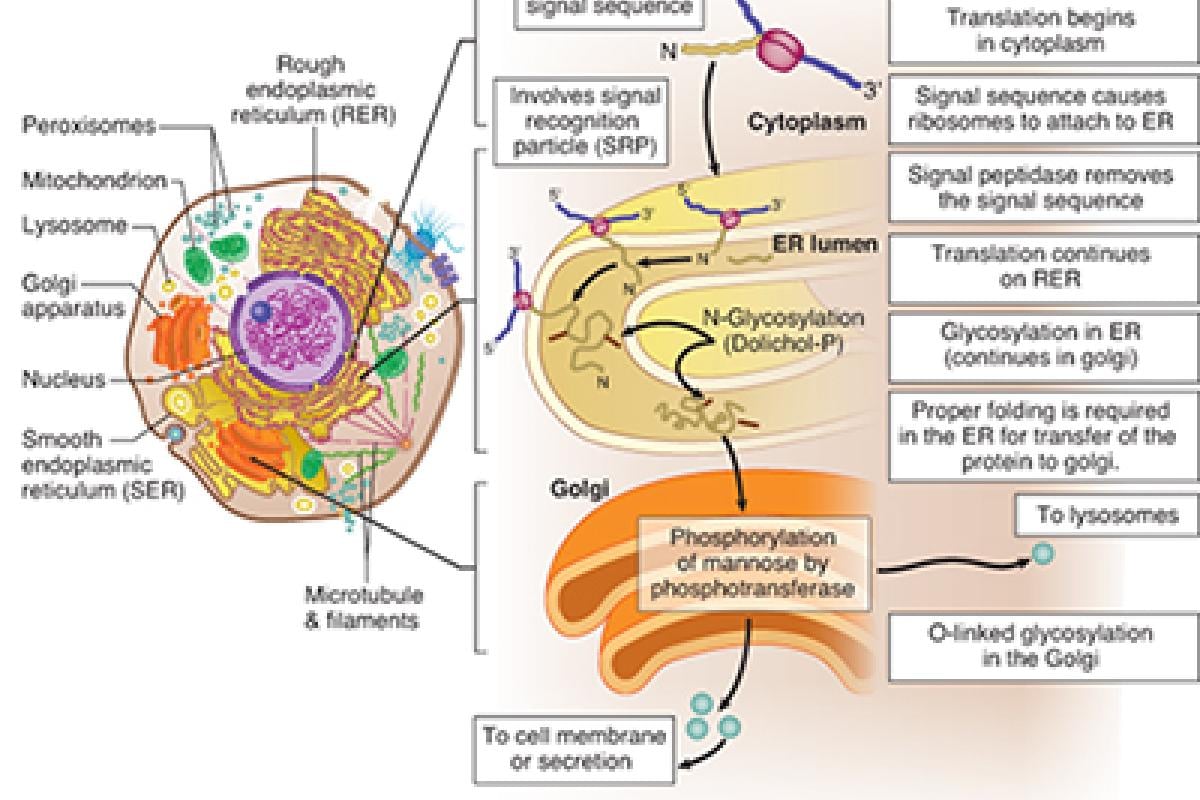
In I-cell disease, this terminal mannose moiety is not phosphorylated, and the acid hydrolases follow the default pathway and are secreted. Like many of the lysosomal storage diseases, the lack of lysosomal enzymes causes structural defects in the cells.
One of the characteristics of I-cell disease is abnormal vacuolization and inclusion bodies in the cytoplasm. By the age of 6 months, there is a failure to thrive (and other developmental delays) as well as abnormal skeletal development, coarse facial features and restricted joint movement.
Why the other answers are wrong
Read these explanations to understand the important rationale for why each answer is incorrect.
Subscribe and succeed in medical school
Get tips and insider advice from the AMA on succeeding in medical school—delivered to your inbox.

Choice A: Hexosaminidase A deficiency (Tay-Sachs disease) is one example of a condition in which ganglioside accumulation occurs.
Choice B: Deficiency of alpha-L-iduronidase results in lysosomal accumulation of dermatan sulfate and heparan sulfate in several conditions, such as mucopolysaccharidosis I, Hurler disease, and Hurler-Scheie syndrome.
Choice C: There are several diseases in which glycogen degradation is defective. These are collectively termed glycogen storage diseases because they result in abnormal cellular accumulation of glycogen. In Pompe disease, or type II glycogen storage disease, a lysosomal glucosidase is deficient, resulting in lysosomal glycogen accumulation.
Choice D: Deficiency of sphingomyelinase, an enzyme involved in degradation of sphingomyelin, results in Niemann-Pick disease.
Choice F: Phosphorylation of tyrosine in tyrosine kinase receptors is unrelated to lysosomes or lysosomal enzymes; however, decreased ability to phosphorylate tyrosine moieties might be associated with diabetes or dwarfism.
Tips to remember
- I-cell disease defect: phosphotransferase activity in the Golgi apparatus.
- Tay-Sachs disease defect: hexosaminidase A activity in the lysosome.
- Niemann-Pick disease defect: sphingomyelinase activity in the lysosome.
- Hurler disease defect: iduronidase activity in lysosome.
The AMA and Kaplan have teamed up to support you in reaching your goal of passing the USMLE® or COMLEX-USA®. If you're looking for additional resources, Kaplan provides free access to tools for pre-clinical studies, including Kaplan’s Lecture Notes series, Integrated Vignettes, Shelf Prep and more.
For more prep questions on USMLE Steps 1, 2 and 3, view other posts in this series..