If you’re preparing for the United States Medical Licensing Examination® (USMLE®) Step 1 exam, you might want to know which questions are most often missed by test-prep takers. Check out this example from Kaplan Medical, and read an expert explanation of the answer. Also check out all posts in this series.
A 20-year-old woman comes to the physician for a routine visit. She has a history of alpha-thalassemia trait and currently does not take any medications. She has had regular menses since she was 12 years old and had average blood loss with each cycle.
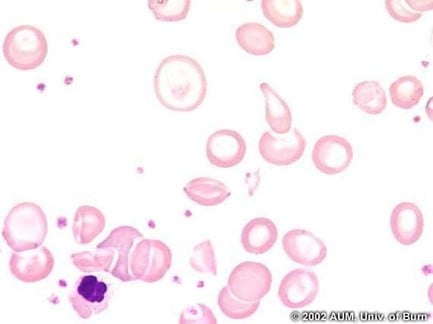
Her vital signs and physical examination are within normal limits. Laboratory studies show leukocyte count of 8,000/mm3, hemoglobin of 11 g/dL, hematocrit of 34%, platelet count of 245,000/mm3, and mean corpuscular volume of 66 μm3. Serum ferritin and red cell distribution width were both within normal limits. The peripheral blood smear is shown.
Which of the following conditions is also associated with the same type of pathologic red blood cell form seen in this patient’s condition?
A. G6PD deficiency.
B. HbC disease.
C. Lead poisoning.
D. Myelofibrosis.
E. Sickle cell disease.
Subscribe and succeed in medical school
Get tips and insider advice from the AMA on succeeding in medical school—delivered to your inbox.
The correct answer is B.
Kaplan Medical explains why
This patient has alpha-thalassemia, which is caused by inherited alpha-globin gene deletions on chromosome 16, leading to decreased alpha-globin chain synthesis, and microcytic, hypochromic anemia with normal serum ferritin levels and red cell distribution width. The amount of gene deletions (out of four total genes) determines the severity of anemia.
Deletion of one gene is not associated with significant anemia, and patients are typically asymptomatic. Two deleted genes (alpha-thalassemia trait) usually present with a mild anemia in asymptomatic patients. Three deleted genes result in HbH disease, characterized by hemoglobin composed of four beta-globin chains, which has a higher affinity for oxygen but is prone to hemoglobin precipitation, leading to membrane damage and extravascular hemolysis. Deletion of all four genes is incompatible with life. Hemoglobin electrophoresis is usually normal in alpha-thalassemia, due to a proportional decrease in all normal hemoglobin types that consist of two alpha-globin chains.
Alpha-thalassemia is associated with target cells on blood smear. Causes of target cells can be memorized by the mnemonic HALT: HbC disease, Asplenia, Liver disease and Thalassemia. HbC disease is caused by a point mutation resulting in glutamate-to-lysine amino acid substitution in beta-globin chains and is also associated with target cells. Sickle cell disease has a similar pathogenesis, but HbC disease is associated with a milder anemia compared to sickle cell disease.
Why the other answers are wrong
Choice A: G6PD deficiency is caused by an X-linked mutation resulting in normocytic, normochromic hemolytic anemia typically induced by oxidant stress from certain exposures, including medications such as sulfa drugs. It is associated with Heinz bodies caused by hemoglobin precipitation, which are extruded by splenic macrophages leading to the presence of bite cells (instead of target cells) on peripheral blood smear.